When cyp2e1 knockout mice that lack the ability to produce the CYP2E1 enzyme were challenged with acetaminophen, they were found to be considerably less sensitive to its hepatotoxic effects than wild-type animals, indicating that this CYP is the principal enzyme responsible for the metabolic conversion of the drug acetaminophen to its active hepatotoxic metabolite NAPQI (Figure 2).[35] During fasting and diabetic ketosis, serum acetone, acetol, and 1,2-propanediol are elevated. CYP2E1 is concomitantly induced due to protein stabilization by acetone.35 Acetone is primarily oxidized to acetol by CYP2E1. As fasting increases ketone production with a concomitant increase in CYP2E1 activity, this in turn increases the production of NAPQI and decreases amino acid substrates for GSH production.[35] Administering acetaminophen is likely to be much more toxic under fasting conditions, such as when a child has an illness that decreases appetite. Children who are sick and fasting and are administered vaccines with prophylactic acetaminophen are much more likely to suffer acetaminophen toxicity.
An examination of the importance of GSH and its biosynthesis is important to an understanding of acetaminophen toxicity. GSH is a tripeptide composed of the amino acids glutamate, cysteine and glycine (Figure 3a). It is present in virtually all aerobic cells at millimolar concentrations where it takes part in numerous fundamental processes. It is a very important antioxidant, participates in detoxification of certain drugs, toxic environmental chemicals, protects against lipid peroxidation and electrophiles, has antiviral effects, is involved in the biosynthesis of DNA, proteins, and leukotrienes, cell proliferation, apoptosis, neurotransmission, and neuromodulation.[58] Decreased levels of GSH are found in several diseases such as liver cirrhosis, pulmonary disease, gastrointestinal or pancreatic inflammation, diabetes, HIV infection, and neurodegenerative diseases.[58] In the normal physiological state (Figure 3a), GSH is produced by the condensation of the amino acids glutamate and cysteine to form the dipeptide γ-glutamylcysteine, which is catalyzed by the enzyme, γ-glutamyl cysteine synthetase.[58] The dipeptide then condenses with the amino acid glycine to form the tripeptide GSH which is catalyzed by the enzyme GSH synthetase. The end product, GSH, exhibits negative feedback inhibition of γ-glutamylsynthetase to prevent the overproduction of GSH. If the amino acid glycine is present at high concentrations, γ-glutamylcysteine is converted in small amounts to pyroglutamic acid that can be recycled to form glutamate.
If GSH is severely depleted by the toxic metabolite of acetaminophen, NAPQI (Figure 3b), and/or there is inadequate glycine to produce GSH due to illness or nutritional deficiency, there is a lack of negative feedback of GSH on γ-glutamylsynthetase.
This severe depletion of GSH results in the synthesis of large amounts of γ-glutamylcysteine, which is increasingly converted to pyroglutamate when glycine is depleted, leading to metabolic acidosis.
As organic acid testing to determine pyroglutamate is usually performed only at specialized pediatric hospitals with specialized biochemical genetics services, the diagnosis of this metabolic disorder in patients may frequently be missed. In addition, NAPQI also deactivates some of the enzymes that recycle GSH, such as GSH peroxidase.[33] The depletion of GSH diminishes the ability of the body to detoxify toxic chemicals. As of 2012, there were 170 articles that indicated an association between toxic chemical exposure and autism.[3] The depletion of GSH will lead to enhanced toxicity of a large number of toxic chemicals. For example, Adams et al. found that variations in the severity of autism measurements could be explained, in part, by regression analyses of urinary excretion of toxic metals before and after DMSA, the chelating agent dimercaptosuccinic acid, and the level of red blood cell glutathione.[59] Thus, partial depletion of GSH by moderate increases in NAPQI could lead to enhanced toxicity of heavy metals and perhaps many other toxic chemicals.
Depletion of GSH as a consequence of acetaminophen toxicity to the liver has attracted the most attention in the medical scientific community, as it can frequently be fatal or require a liver transplant or emergency treatment to prevent liver failure (the liver is the organ with the greatest concentration of GSH). However, acetaminophen toxicity has been implicated in a wide range of other disorders in humans and/or experimental animals including cancer, birth defects, asthma, allergies, and brain toxicity (Table 1).
Concentrations of p-aminophenol (which is converted to the cannabinoid substance AM404; Figure 2) from 1 to 100 μg/mL produced significant loss of mouse cortical neuron viability at 24 h compared with the controls.11 The naturally occurring endocannabinoid anandamide also caused similar 24 h loss of cell viability in developing mouse cortical neurons at concentrations ranging from 1 to 100 μg/mL, indicating the mechanism of cell death could be mediated through the cannabinoid receptors. Defective glucuronidation would also increase the conversion of acetaminophen to its more toxic metabolites, NAPQI and AM 404. Such defects in glucuronidation are common in the developing fetus and in newborns.[54]
Posadas et al. found that acetaminophen causes concentration-dependent neuronal death in vitro at concentrations that are reached in human plasma during acetaminophen overdose, and are reached in the cerebrospinal fluid of rats for 3 h following doses that are below those required to induce acute hepatic failure in rats.[15] Acetaminophen also increases both neuronal CYP2E1 enzymatic activity and protein levels, as determined by western blot, leading to neuronal death through mitochondrialmediated mechanisms that involve cytochrome c release and caspase 3 activation. In addition, in vivo experiments show that acetaminophen injection induces neuronal death in the rat cortex. Posadas et al. established a direct neurotoxic action by acetaminophen both in vitro and in vivo in rats at doses below those required to produce hepatotoxicity and suggested that this neurotoxicity might be involved in the general toxic syndrome observed during patient acetaminophen overdose and, possibly, when acetaminophen doses in the upper dosing schedule are used, especially if other risk factors (moderate alcohol drinking, fasting, nutritional impairment) are present.[15]
Purkinje Cell Abnormalities, Autism, and GSH Depletion
Ritvo et al. were the first to report abnormalities of Purkinje cells in the cerebella of people with autism.[60] These cells are some of the largest neurons in the human brain, with an intricately elaborate dendritic arbor, characterized by a large number of dendritic spines; these neurons utilize γ-aminobutyric acid as their neurotransmitter and send inhibitory projections to the deep cerebellar nuclei, and constitute the sole output of all motor coordination in the cerebellar cortex.[61] The initial study of Ritvo et al. indicated that the number of Purkinje cells in the vermis of the cerebellum was 15 standard deviations below the mean and approximately 8 standard deviations below the mean bilaterally in the cerebral hemispheres of the individuals with autism compared with controls. Vargas et al. found that brain tissues of autistic patients showed extensive neuroglial responses characterized by microglial and astroglial activation.[62] In the brains of autistic patients, the most prominent histological changes were observed in the cerebellum, characterized by a patchy loss of neurons in the Purkinje cell layer and granular cell layer in nine out of ten cerebella; one of these cerebella also showed an almost complete loss of Purkinje cells from the Purkinje cell layer, as well as a marked loss of granular cells. Kern and Jones have summarized the important role of Purkinje cell abnormalities in autism, especially the susceptibility of these cells to oxidative stress during GSH depletion.[63] Such depletion can be due to reduction of GSH due to excessive NAPQI exposure and/or to GSH depletion associated with elevated dopamine secondary to dopamine β-hydroxylase inhibition by phenolic Clostridia metabolites. The metabolite of acetaminophen, 4-amino phenol, also caused depletion of GSH.[64]
Immune Abnormalities Associated with Acetaminophen Use
An important but under-appreciated aspect of acetaminophen toxicity is that direct, drug-induced harm accounts for only part of the overall syndrome of acetaminophen-induced liver injury. The reason for this is that the initial wave of drug-induced hepatocellular destruction is followed by a robust innate immune response, in which invading inflammatory cells release toxic oxidants and cause a second wave of destruction. The collateral damage inflicted by inflammatory cells can be so severe as to double the degree of tissue injury caused by acetaminophen alone.[34]
Prymula and colleagues compared post-vaccine fever and post-vaccine changes in vaccine-specific antibody titers in children who either did or did not receive scheduled acetaminophen with immunizations.[37] Four hundred and fifty nine healthy infants undergoing initial and booster vaccinations for a variety of vaccines were randomized to receive or not to receive prophylactic acetaminophen at the time of and for 24 h after vaccinations. Fever was significantly less common in acetaminophen-treated children after initial and booster vaccination. Children who received acetaminophen had significantly lower antibody titers to each vaccine-related antigen, although it was judged that the titers would probably be protective against the diseases for which they had been immunized. In children with severe sulfation deficiency with a predisposition to autism, vaccination without an adequate immune response might lead to viral infection even with the attenuated strains of the viral vaccines due to a lack of GSH needed for effective immune response.[46]
Acetaminophen sales were high in Englishspeaking countries, and were positively associated with asthma symptoms, eczema and allergic rhinoconjunctivitis in children aged 13–14, and with wheeze, diagnosed asthma, rhinitis and bronchial responsiveness in adults. The prevalence of wheeze increased by 0.52% in 13–14 year olds and by 0.26% in adults (p < 0.0005) for each gram increase in per capita acetaminophen sales. Between 1980 and 2003, the prevalence of pediatric asthma in the United States increased from 3.6% to 5.8%, and similar increases were observed throughout the world.[45] It has been speculated that frequent use of acetaminophen might influence asthma and rhinitis by depleting levels of reduced GSH in the nose and airways, thus shifting the oxidant/antioxidant balance in favor of oxidative stress and increasing inflammation.[44]
Acetaminophen decreases GSH levels, principally in the liver and kidneys, but also in the lungs.[46] These decreases are dose dependent; overdose levels of acetaminophen are cytotoxic to pneumocytes and cause acute lung injury, whereas nontoxic, therapeutic doses produce smaller, but significant reductions in GSH levels in type II pneumocytes and alveolar macrophages.[46] Among healthy young volunteers, significantly lower serum antioxidant capacity has been seen within 2 weeks of ingestion of 1 g of acetaminophen.[46] By depleting GSH levels, acetaminophen weakens the ability of the host to mitigate oxidative stress produced by reactive oxygen species (ROS) such as superoxide anions (O2 −), hydroxyl (•OH), and peroxyl (ROO−) radicals.[46]Finally, when GSH levels are low, defective processing of disulfide bonds that are key in antigen presentation has been hypothesized.[46] It is conceivable that decreased levels of GSH guide the expression of T-helper cell pathways by altering antigen presentation and recognition, thereby favoring the T2 allergic-dominant pathway. In a study of children with autism spectrum disorders (ASDs) in Sweden, airway symptoms of wheezing and physician-diagnosed asthma in the baseline investigation in infants and toddlers were associated with ASD 5 years later.[52]
Skewed T1 or T2 responses were also indicated in ASD children.[65, 66] Analysis by Jyonouchi et al. of adaptive immune responses revealed markedly variable T1rT2 cytokine levels in ASD children compared with control siblings.[67] It has been reported that immune responses in autistic children are relatively skewed to T2 on the basis of intracellular staining of T1rT2 cytokines.[65] Higher levels of IgG, IgA, and IgE allergies to various foods, but especially milk and wheat, have been reported in children with autism, together with abnormal cellular immune responses to milk, wheat, and soy.[67] It has been shown that depleting GSH in brain microglia and astroglia induces a neuroinflammatory response that results in both significant cytokine release and the release of material that is toxic to neurons.
In addition, the ability of GSH to downregulate nuclear factor-kB, and the inverse association between alveolar GSH levels and bronchial responsiveness, suggests that GSH may modify asthma inflammation. Secondly, studies in animals have found that acetaminophen can deplete the lung of GSH. These effects in macrophages raise the possibility that acetaminophen might also influence atopic diseases more generally through another mechanism, namely the promotion of atopy, since depletion of GSH in antigen-presenting cells promotes T-helper cell 2-type cytokine responses. This might explain why, in children, acetaminophen sales were associated with atopic eczema as well as with asthma and rhinitis. As of 2012, 416 studies had demonstrated an association between autism and immune abnormalities and/or inflammation.[3]
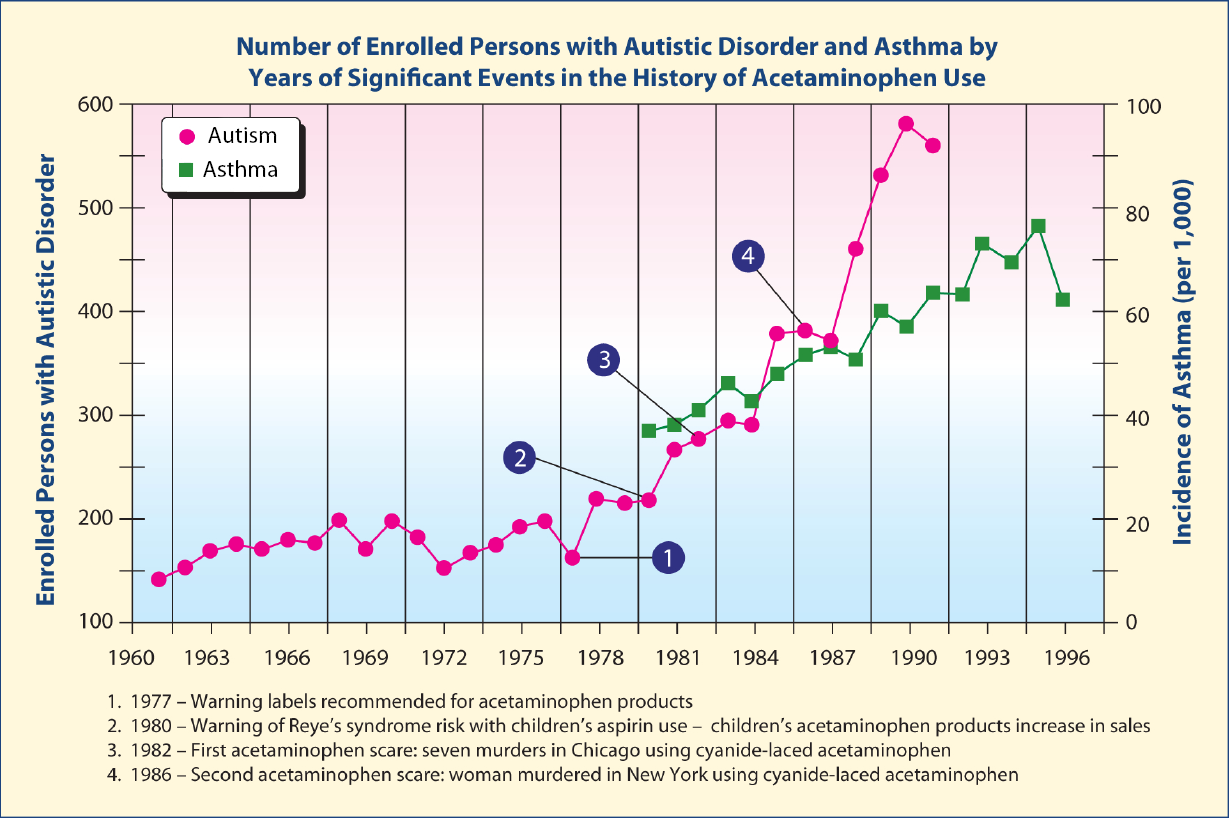
The beginning of the rapid increase in autism in around 1980 coincides with the rapid increase in asthma, both of which coincide with the rapid increase in the use of acetaminophen following the Reye's syndrome scare over a possible association with aspirin.
It would seem likely that perhaps some children on the autistic spectrum might have both brain and lung abnormalities caused by acetaminophen. Is there any evidence for such combined abnormalities? Indeed, at the annual meeting of the American College of Chest Physicians in Honolulu, Hawaii in 2011, Barbara Stewart, a pediatric pulmonologist, found that a significant lung abnormality was present in 100% of children (n = 47) on the autistic spectrum who were examined, but in none of <300 children without autism.68 Most of the children with autism had been referred to her clinic due to persistent cough unresponsive to treatment. She noticed during bronchoscopy examinations, in which a lighted tube is inserted into the lungs, that, although the airways of the children initially appeared normal, the lower airway had doubled branches, or "doublets". Dr. Stewart said "when airways divide beyond the first generation, they typically branch like a tree, with one branch on one side and one on the other. A doublet occurs when there are twin branches that come off together instead of one, which are exactly symmetrical, in each of the lower locations that can be seen." In a study of ASD children in Sweden, airway symptoms of wheezing and physician-diagnosed asthma in the baseline investigation in infants and toddlers were associated with ASD 5 years later.[52] It would seem very useful to examine the use of acetaminophen both prenatally and postnatally in the children with the abnormal lung anatomy.
DNA Damage
As of 2012, 145 studies had shown an association between mitochondria function and autism.[3] The data of Cover et al. showed that nitration of mitochondrial proteins and depletion of mitochondrial DNA after acetaminophen overdose in mice was due to peroxynitrite formation.[32] In contrast, nDNA damage was not directly caused by peroxynitrite. Nuclear DNA damage after acetaminophen overdose is likely to be caused by DNase(s) unrelated to the caspase-activated DNase, which is typically responsible for DNA fragmentation during apoptosis. The data from Cover et al. suggest that the activation of these DNase(s) is dependent on the mitochondrial oxidant stress and peroxynitrite formation.[32]
Recent data have demonstrated that nitrated tyrosine residues, as well as acetaminophen adducts, occur in the necrotic cells following toxic doses of acetaminophen. Nitrotyrosine was postulated to be mediated by peroxynitrite, a reactive nitrogen species formed by the very rapid reaction of superoxide and nitric oxide (NO). Peroxynitrite is normally detoxified by GSH, which is depleted in acetaminophen toxicity. NO synthesis (serum nitrate plus nitrite) was dramatically increased following acetaminophen.
NAPQI
The metabolism of acetaminophen (Figure 2) by CYP2E1 forms NAPQI, a reactive metabolite that binds to cysteine residues in cellular proteins and forms acetaminophen protein adducts, referred to as acetaminophen-cysteine complexes (APAP-CYS).
Heard et al. demonstrated that low concentrations of APAP-CYS are detectable in serum following therapeutic dosing with acetaminophen in the vast majority of individuals.[49] The APAP-CYS concentrations following acetaminophen overdose varied widely. Serum APAP-CYS concentrations in patients with hepatic injury following acetaminophen overdose were generally much higher than those observed during therapeutic dosing. However, three overdose patients had APAP-CYS concentrations that were of the same order of magnitude as those observed with therapeutic dosing. Adduct concentrations varied according to the degree of exposure. Currently, an absolute adduct level exceeding 1.1 µmol/L appears consistent with acetaminophen toxicity. While therapeutic dosing generally produces concentrations below 0.5 µmol/L, values in people taking recommended dosages have been reported to be very close to the lower limit of APAP-CYS concentration associated with acetaminophen toxic overdoses (1.0 µmol/L).
NAPQI and Anomalous Hair Metal Values in Autism
Concentrations of the NAPQI metabolite increased to values as high as 1.0 µmol/L or 1000 nmol/L serum following therapeutic doses of acetaminophen. Hair proteins contain a considerable amount of cysteine, and hair proteins in hair follicles and would be expected to react with NAPQI to form NAPQI adducts with cysteine sulfhydryl groups in hair. Such cysteine groups also react strongly with heavy metals such as mercury. In comparison, the mean blood mercury concentration of children with autism was 19.53 nmol/L. Thus the concentration of NAPQI might be as much as 51 times the concentration of total mercury. Since NAPQI amounts are so much higher than mercury values, it would be expected that NAPQI might significantly reduce the capacity of hair proteins to bind mercury if the binding capacity of the hair sulfhydryl groups was exceeded by the previous binding of NAPQI. Such inhibition by NAPQI may help to explain anomalous data in the measurement of mercury in hair samples of children with autism. In two baby hair studies in which samples were obtained at first haircuts, mercury values in hair were much lower in children with autism compared with those of normal controls.[69, 70] In addition, children with the most severe symptoms of autism had the lowest amount of hair mercury. Studies in Kuwait and Saudi Arabia found opposite results in children with autism; children with autism had much higher amounts of hair mercury as well as other heavy metals, but in both of these studies the children were much older (mean ages 8.8 years in Saudi Arabia and 4.2 years in Kuwait) than those in the baby hair studies (12–24 months of age).[71, 72] All of this data could be explained by patterns of acetaminophen drug use in children. Acetaminophen is commonly used prophylactically to prevent fever in infants and toddlers who receive the bulk of their vaccines in the first 2 years of life. In contrast, few vaccines and attendant prophylactic acetaminophen are administered to older children with autism in the age range in which hair mercury values were elevated compared with controls. In these older children, hair metals such as mercury would be considerably higher due to a lack of competition from NAPQI for reactive sulfhydryl groups in hair proteins in the hair follicles.
Special Concerns About the Long-Term Defective Quality Control of Acetaminophen Products
All of the information to this point has been directed to the toxicity of pure acetaminophen. Toxicity is usually due to exceeding the recommended daily dosage, often occurring due to accidental combination use of multiple products containing acetaminophen or through changes in dosage amounts by manufacturers. Adding to this are reports that acetaminophen products have been contaminated at various times, potentially increasing health risks. These problems have been documented in Food and Drug Administration inspection reports and subsequent recalls.
From the time it was introduced in 1955, acetaminophen has become one of the most successful OTC drugs, and has earned the primary manufacturer, Johnson & Johnson, an estimated US$1.3 billion every year.[73]Within the last 10 years, the FDA has chronicled manufacturing problems including mislabeling of children's tablets, where packages of the product listed an incorrect amount of the ingredients per tablet, inadequate cleaning of equipment used for manufacturing, insufficient follow-up in investigating consumer complaints, product mix-ups, and contamination with metal fragments and bacteria.[74, 75] The latter resulted in a recall of 135 million bottles of children's medications containing acetaminophen.
Additional recalls occurred in 2009 and 2010 due to trace amounts of the chemical 2,4,6-tribromoanisole found in infant, children, and adult OTC products. This recall was prompted by 775 consumer reports of nausea, vomiting, stomach pains and diarrhea received by FDA due to the contamination.[76] It was found that the chemical 2,4,6-tribromoanisole (a metabolite of a chemical fungicide tribromophenol) was being used in a manufacturing facility to treat the wood used in pallets used for transporting packaged materials.[77]Tribromoanisole is produced when naturally-occurring airborne fungi and/or bacteria (usually Aspergillus sp.,Penicillium sp., Actinomycetes, Botrytis cinerea, Rhizobium sp., or Streptomyces) are presented with brominated phenolic compounds, which they then convert into bromoanisole derivatives.
Other FDA-publicized inspection reports of acetaminophen-manufacturing facilities have listed inadequate quality controls, lack of safeguards to isolate "rejected" raw materials and other drugs, and several human errors resulting in product mix-ups.[78] Several recalls have occurred due to failed FDA manufacturing inspections and contaminated products affecting >300 million bottles of adult and children's medicines.[79, 82] Manufacturing controls are an integral aspect of pharmaceutical production to ensure public safety, and disregard or deficiency can lead to serious outcomes. In response to the high number of inadequacies found through inspections and numerous recalls, US health authorities have taken over supervision of three acetaminophen manufacturing plants to mitigate risks.[83]
A Call to Action
A large-scale, long-term study of both prenatal and postnatal effects of acetaminophen exposure on the incidence of autism, attention deficit with hyperactivity, and asthma should be conducted. However, such a study would probably take at least 5 years. If acetaminophen is indeed the cause of all of these illnesses, should acetaminophen use continue for such a long period of time while millions of additional children are further affected? Respected physicians consider that the connection of acetaminophen with asthma has been proven beyond a reasonable doubt. Dr McBride, Professor of Pediatrics at Department of Pediatrics, Northeast Ohio Medical University, Rootstown, Ohio summarizes the evidence for the acetaminophen asthma findings[45]: "There remains a possibility that confounding variables might explain some or all of the association between acetaminophen and asthma. For this reason we need further studies. At present, however, I need further studies not to prove that acetaminophen is dangerous but, rather, to prove that it is safe. Until such evidence is forthcoming, I will recommend avoidance of acetaminophen by all children with asthma or those at risk for asthma and will work to make patients, parents, and primary care providers aware of the possibility that acetaminophen is detrimental to children with asthma."
Since all children may be at risk from asthma, Dr. McBride is in effect saying that acetaminophen is contraindicated for the treatment of any children. Although the case for acetaminophen being a cause of autism and attention deficit with hyperactivity may not be as strong as the case for asthma, the severe asthma risk combined with the risks of autism and attention deficit with hyperactivity are so severe that we as a society should maintain a degree of caution with acetaminophen given the proven overall toxicity due to accidental overdose of the drug, and the availability of ibuprofen or abstaining from treatment as alternatives. A large-scale trial of acetaminophen restriction in pregnancy and the first 3 years of life is warranted to test the hypothesis that acetaminophen is a causative agent in autism, asthma, and attention deficit with hyperactivity. Due to the increased risks associated with accidental overuse of acetaminophen, increased public awareness of such risks is paramount.
Given associations of acetaminophen with increased rates of cancer, increased testicular damage, increased rates of asthma, and allergy, plausible causation of autism, and in vitro evidence of brain damage associated with metabolites of acetaminophen, new assessments of the relative risk of aspirin causing Reye's syndrome versus the risks of acetaminophen in children should be undertaken. If a clear link between acetaminophen use pre- or post-natally and autism is established, medical practice guidelines may need to be adjusted and alternative analgesics or antipyretics, such as ibuprofen, recommended. However, such a relationship may be difficult to establish, as studies may have to account for variations in dosage amounts, formats, combination product use, and for the possibility of product recalls given the recent reported manufacturing issues surrounding acetaminophen.
Note Added in Proof
After this article was submitted for publication on January 2, 2013 the following article was published prior to publication which strongly supports the hypothesis in the current article.
In 2013, Bauer and Kriebel reported that prenatal use of acetaminophen was strongly correlated with autism/Autism Spectrum Disorder prevalence (r = 0.80) using all available country-level data (n = 8) for the period 1984 to 2005. In addition, the authors found that after acetaminophen became commonly used to treat circumcision pain after 1995, there was a strong correlation between countrylevel (n = 9) autism/ASD prevalence in males and a country's circumcision rate (r = 0.98). A very similar pattern was seen among US states and when comparing the three main racial/ethnic groups in the US.[84]
Clinical References:
Kim YS, Leventhal BL, Koh YJ, Fombonne E, Laska E, Lim EC, et al. Prevalence of autism spectrum disorders in a total population sample. Am J Psychiatry. 2011;168(9):904–12.
Hertz-Picciotto I, Delwiche L. The rise in autism and the role of age at diagnosis. Epidemiology. 2009;20(1):84–90.
Rossignol DA, Frye RE. A review of research trends in physiological abnormalities in autism spectrum disorders: Immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol Psychiatry. 2012;17(4):389–401.
Shaw W. The unique vulnerability of the human brain to toxic chemical exposure and the importance of toxic chemical evaluation and treatment in orthomolecular psychiatry. J Orthomol Med. 2010;25(3):125–34.
Miller MT. Thalidomide embryopathy: A model for the study of congenital incomitant horizontal strabismus. Trans Am Ophthalmol Soc. 1991;89:623–74.
Disability-analysis of State Reports, reviewed by the Committee on the Rights of the Child (CRC) in its Pre- Sessional Working Group, 55th Session. International Disability Alliance 2010;8.
http://www.autism-society.org/about-autism/. Autism Society Website 2012. Accessed August 31, 2013.
Reed G, Galindo MA. Cuba's National Immunization Program. MEDICC Review. 2008;9(1):5–7.
Bäckström M, Hägg S, Mjörndal T, Dahlqvist R. Utilization pattern of metamizole in northern Sweden and risk estimates of agranulocytosis. Pharmacoepidemiol Drug Saf. 2002;11(3):239–45.
Torres AR. Is fever suppression involved in the etiology of autism and neurodevelopmental disorders? BMC Pediatr. 2003;2:3–9.
Schultz ST, Klonoff-Cohen HS, Wingard DL, Akshoomoff NA, Macera CA, Ji M. Acetaminophen (paracetamol) use, measles-mumps rubella vaccination, and autistic disorder: The results of a parent survey. Autism. 2008;12(3):293–307.
Schultz ST. Can autism be triggered by acetaminophen activation of the endocannabinoid system? Acta Neurobiol Exp. 2010;70(2):227–31.
Schultz S, Desilva M, Gu TT, Qiang M, Whang K. Effects of the analgesic acetaminophen (paracetamol) and its para-aminophenol metabolite on viability of mouse-cultured cortical neurons. Basic Clin Pharmacol Toxicol. 2011;110(2):141–4.
Becker KG, Schultz ST. Similarities in features of autism and asthma and a possible link to acetaminophen use. Med Hypotheses. 2010;74(1):7–11.
Posadas I, Santos P, Blanco A, Muñoz-Fernández M, Ceña V. Acetaminophen induces apoptosis in rat cortical neurons. PLoS ONE. 2010;15(12):e15360.
U.S. Food and Drug Administration. For consumers – Don't double up on acetaminophen. 2013. Available at:http://www.fda.gov/ForConsumers/ConsumerUpdates/ucm336581.htm . Accessed March 19, 2013.
Connor DF. Problems of overdiagnosis and overprescribing in ADHD. Psychiatric Times. 2011;28(8):1–8.
Center For Disease Control. Mental health in the United States: Prevalence of diagnosis and medication treatment for attention-deficit/hyperactivity disorder – United States, 2003. MMWR. 2005;54(34):842–7.
Safer DJ. Are stimulants overprescribed for youths with ADHD? Ann Clin Psychiatry. 2000;12(1):55–62.
Safer D, Krager JM. Hyperactivity and inattentiveness. School assessment of stimulant treatment. Clin Pediatr. 1989;28(5):216–21.
Safer DJ, Zito JM, Fine EM. Increased methylphenidate usage for attention deficit disorder in the 1990s. Pediatrics. 1996;98(6):1084–8.
Safer DJ, Krager JM. The increased rate of stimulant treatment for hyperactive/inattentive students in secondary schools.Pediatrics. 1994;94(4):462–4.
Safer DJ, Malever M. Stimulant treatment in Maryland Public Schools. Pediatrics. 2009;106(3):533–9.
Safer DJ, Krager JM. A survey of medication treatment for hyperactive/inattentive students. J Am Med Assoc. 1988;260(15):2256–8.
Mandell DS, Thompson WW, Weintraub ES, Destefano F, Blank MB. Trends in diagnosis rates for autism and ADHD at hospital discharge in the context of other psychiatric diagnoses. Psychiatr Serv. 2005;56(1): 56–62.
Orlowski JP, Hanhan UA, Fiallos MR. Is aspirin a cause of Reye's syndrome? A case against. Drug Saf. 2002;25(4):225–31.
Van Bever HP, Quek SC, Lim T. Aspirin, Reye syndrome, Kawasaki disease, and allergies; a reconsideration of the links. Br Med J. 2004;89(12):1178.
Weiner DL. Reye Syndrome. 2011. Available at: http://EMedicine.Medscape.com. Accessed August 31, 2013.
Shaw W. Possible role of lysolecithins and nonesterified fatty acids in the pathogenesis of Reye's syndrome, sudden infant death syndrome, acute pancreatitis, and diabetic ketoacidosis. Clin Chem. 1985;31(7):1109–15.
Kristensen DM, Hass U, Lesné L, Lottrup G, Jacobsen PR, Desdoits-Lethimonier C, et al. Intrauterine exposure to mild analgesics is a risk factor for development of male reproductive disorders in human and rat. Human Reprod.2010;26(1):235–44.
Adjei AA, Gaedigk A, Simon SD, Weinshilboum RM, Leeder JS. Interindividual variability in acetaminophen sulfation by human fetal liver: Implications for pharmacogenetic investigations of drug-induced birth defects. Birth Defects Res A Clin Mol Teratol. 2008;82(3):155–65.
Cover C, Mansouri A, Knight TR, Bajt ML, Lemasters JJ, Pessayre D, et al. Peroxynitrite-induced mitochondrial and endonuclease-mediated nuclear DNA damage in acetaminophen hepatotoxicity. J Pharmacol Exp Ther. 2005;315(2):879–87.
Ruepp SU, Tonge RP, Shaw J, Wallis N, Pognan F. Genomics and proteomics analysis of acetaminophen toxicity in mouse liver. Toxicol Sci. 2012;65(1):135–50.
Maher JJ. DAMPs ramp up drug toxicity. J Clin Invest. 2009;119(2):246–9.
Lee SS, Buters JT, Pineau T, Fernandez-Salguero P, Gonzalez FJ. Role of CYP2E1 in the hepatotoxicity of acetaminophen.J Biol Chem. 1996;271(20):12063–7.
Yamaura K, Ogawa K, Yonekawa T, Nakamura T, Yano S, Ueno K. Inhibition of the antibody production by acetaminophen independent of liver injury in mice. Biol Pharm Bull. 2002;25(2):201–5.
Prymula R, Siegrist CA, Chlibek R, Zemlickova H, Vackova M, Smetana J, et al. Effect of prophylactic paracetamol administration at time of vaccination on febrile reactions and antibody responses in children: Two open-label, randomised controlled trials. Lancet. 2009;374:1339–50.
Homme JH, Fischer PR. Prophylactic paracetamol at the time of infant vaccination reduces the risk of fever but also reduces antibody response. Evid Based Med. 2010;15(2):50–1.
Fenves AZ, Kirkpatrick III HM, Patel VV, Sweetman L, Emmett M. Increased anion gap metabolic acidosis as a result of 5-oxoproline (pyroglutamic acid): A role for acetaminophen. Clin J Am Soc Nephrol. 2006;1(3):441–7.
Pitt JJ, Brown GK, Clift V, Christodoulou J. Atypical pyroglutamic aciduria: Possible role of paracetamol. J Inherit Metab Dis. 1990;13(5):755–6.
Miller AL. Liver damage from chronic acetaminophen dosing is dangerous, but not the only risk. Alt Med Rev. 2009;14(4):322–3.
Franciscus A, Highleyman L. Acetaminophen and your liver. HCSP Fact Sheet. 2009;2.2:1–3.
Walter RB, Milano F, Brasky TM, White E. Long-term use of acetaminophen, aspirin, and other nonsteroidal anti-inflammatory drugs and risk of hematologic malignancies: results from the prospective vitamins and lifestyle (VITAL) study.J Clin Oncol. 2011;94:2424–2431.
Newson RB, Shaheen SO, Chinn S, Burney PG. Paracetamol sales and atopic disease in children and adults: An ecological analysis. Eur Respir J. 2000;16(5):817–23.
McBride JT. Association of acetaminophen and asthma prevalence and severity. Pediatrics. 2011;128(6):1181–5.
Eneli I, Sadri K, Camargo C Jr, Barr RG. Acetaminophen and the risk of asthma: The epidemiologic and pathophysiologic evidence. Chest. 2005;127(2):604–12.
Somers GF. Pharmacological properties of Thalidomide (a-phthalimido glutarimide), a new sedative hypnotic drug. Br J Pharmacol Chemother. 1960;15:111–116.
Good P. Did acetaminophen provoke the autism epidemic? Alt Med Rev. 2009;14(4):364–72.
Heard KJ, Green JL, James LP, Judge BS, Zolot L, Rhyee S, et al. Acetaminophen-cysteine adducts during therapeutic dosing and following overdose. BMC Gastroenterol. 2011;11:20.
Krakowiak P, Walker CK, Bremer AA, Baker AS, Ozonoff S, Hansen RL, et al. Maternal metabolic conditions and risk for autism and other neurodevelopmental disorders. Pediatrics. 2012;129(5):e1121–8.
Windham GC, Zhang L, Gunier R, Croen LA, Grether JK. Autism spectrum disorders in relation to distribution of hazardous air pollutants in the San Francisco Bay area. Environ Health Perspect. 2006;114(9):1438–44.
Larsson M, Weiss B, Janson S, Sundell J, Bornehag CG. Associations between indoor environmental factors and parental-reported autistic spectrum disorders in children 6–8 years of age. Neurotoxicology. 2009;30(5):822–31.
Kim SJ, Lee MY, Kwon do Y, Kim SY, Kim YC. Alteration in metabolism and toxicity of acetaminophen upon repeated administration in rats. J Pharmacological Sci. 2009;111(2):175–81.
Alcorn J, McNamara PJ. Pharmacokinetics in the newborn. Adv Drug Deliv Rev. 2003;55(5):667–86.
Högestätt ED, Jönsson BA, Ermund A, Andersson DA, Björk H, Alexander JP, et al. Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system. J Biol Chem. 2005;280(36):31405–12.
O'Reilly BA, Waring RH. Enzyme and sulphur oxidation deficiencies in autistic children with knownfood/chemical intolerances. J Orthomol Med. 1993;8(4):198–200.
Alberti A, Pirrone P, Elia M, Waring RH, Romano C. Sulfation deficit in "low functioning" autistic children: A pilot study. Biol Psychiatry. 1999;46(3):420–4.
Schlune A, Mayatepek E. Glutathione synthetase deficiency – An inborn error of the gamma-glutamyl cycle. J Pediatr Sci.2011;3(70):2505–9.
Adams JB, Baral M, Geis E, Mitchell J, Ingram J, Hensley A, et al. The severity of autism is associated with toxic metal body burden and red blood cell glutathione levels. J Toxicol. 2009;2009:532640.
Ritvo ER, Freeman BJ, Scheibel AB, Duong T, Robinson H, Guthrie D, et al. Lower Purkinje cell counts in the cerebella of four autistic subjects: Initial findings of the UCLA-NSAC autopsy research reports. Am J Psychiatry. 1986;143(7):862–6.
Purves D, Augustine GJ, Fitzpatrick D, Hall WC, LaMantia AS, McNamara JO, et al. Neuroscience, 4th edn. Sunderland, MA: Sinaur Associates; 2008:432–4.
Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57(1):67–81.
Kern JK, Jones AM. Evidence of toxicity, oxidative stress, and neuronal insult in autism. J Toxicol. Environ Health B Crit Rev. 2006;9(6):485–99.
Fu X, Chen TS, Ray MB, Nagasawa HT, Williams WM. p-Aminophenol-induced hepatotoxicity in hamsters: Role of glutathione. J Biochem Mol Toxicol. 2004;18(3):154–61.
Gupta S, Aggarwal S, Rashanravan B, Lee T. Th1- and Th2-like cytokines in CD4+ and CD8+ T cells in autism. J Neuroimmunol. 1998;85(1):106–9.
Singh VK. Plasma increase of interleukin-12 and interferon-gamma. Pathological significance in autism. J Neuroimmunol. 1996;66(1-2):143–5.
Jyonouchi H, Sun S, Le H. Proinflammatory and regulatory cytokine production associated with innate and adaptive immune responses in children with autism spectrum disorders and developmental regression. J Neuroimmunol. 2001;120(1–2):170–9.
Harrison L. Autism linked to unusual shapes in lungs study shows children born with a unique airway shape may have autism. WebMD Health News. 2011. Available at: http://www.webmd.com/brain/autism/news/20111025/autism-linked-to-unusual-airwayshapes. Accessed August 31, 2013.
Holmes AS, Blaxill MF, Haley BE. Reduced levels of mercury in first baby haircuts of autistic children. Int J Toxicol. 2003;22(4):277–85.
Adams JB, Romdalvik J, Levine KE, Hu LW. Mercury in first-cut baby hair of children with autism versus typically-developing children. Toxicol Environ Chem. 2008;90(4):739–53.
Fido A, Al-Saad S. Toxic trace elements in the hair of children with autism. Autism. 2005;9(3):290–8.
Al-Ayadhi LY. Heavy metals and trace elements in hair samples of autistic children in central Saudi Arabia. Neurosciences. 2005;10(3):213–8.
Easton T, Herrera S. Despite bad publicity and costly legal settlements, Johnson & Johnson refuses to put ample warnings on its Tylenol labels. Torah View 2012. Available at:https://groups.google.com/forum/#!topic/alt.med.fibromyalgia/grdcWoHo5Hw. Accessed April 26, 2012.
Kavilanz P. Tylenol plant: From bad to worse. CNN Money 2010. Available at:http://money.cnn.com/2010/06/22/news/companies/tylenol_plant_inspection_history/index.htm. Accessed April 26, 2012.
Kavilanz P. Tylenol plant still plagued by FDA violations. CNN Money 2012. Available at:http://money.cnn.com/2010/12/01/news/companies/tylenol_plant_new_problems/index.htm. Accessed April 26, 2012.
Kavilanz P. Tylenol recall: Serious side effects investigated. CNN Money 2010. Available at:http://money.cnn.com/2010/05/25/news/companies/tylenol_recall_adverse_consumer_complaints/index.htm. Accessed April 26, 2012.
Kavilanz P. New recalled Tylenol linked to shut plant. CNN Money 2010. Available at:http://money.cnn.com/2010/10/19/news/companies/new_tylenol_recall/index.htm. Accessed April 26, 2012.
Baker SL. Drugs like Tylenol can be contaminated with mold and chemicals. NaturalNews Com 2010. Available at:http://www.naturalnews.com/027965_Tylenol_recall.html. Accessed April 26, 2012.
Heavey S. FDA finds grime at J&J plant, urges use of generics. Reuters 2010. Available at:http://www.reuters.com/article/2010/05/05/us-johnsonandjohnson-recall-idUSTRE64367Z20100505. Accessed April 26, 2012.
Silverman E. Which Drugmaker Fails Most FDA Inspections? Pharmalot com 2011. Available at: https://seekingalpha.com/article/256209-which-drugmaker-fails-most-fda-inspections. Accessed April 26, 2012.
Krauskopf L. More Tylenol Recalled. The Huffington Post 2011. Available at:http://www.huffingtonpost.com/2011/03/30/tylenol-recalled-_n_842450.html. Accessed April 27, 2012.
Kavilanz P. U.S. takes over three Tylenol plants. CNN Money 2011. Available at:http://money.cnn.com/2011/03/10/news/companies/johnson_mcneil_fda_action/index.htm. Accessed April 27, 2012.
Bauer AZ, Kriebel D. Prenatal and perinatal analgesic exposure and autism: an ecological link. Environ Health. 2013;12:41. doi: 10.1186/1476-069X-12-41.